
无机纳米材料通过催化作用驱动细胞活性氧(H2O2,O2??,O2等)发生化学转化,是其毒性等生物学效应的重要来源,由此开展抗菌、抗氧化、抗肿瘤等生物应用是纳米医学的重要课题。高兴发课题组长期用理论与模拟手段研究纳米材料催化活性氧转化的机制与规律,发展纳米毒性预测理论和相关生物应用的计算设计方法,最近在该方向取得了系列进展。
1. “透明”机器学习虚拟筛选靶向肿瘤H2O2的2D纳米材料
实体瘤富含H2O2分子,纳米材料催化H2O2攫氢反应(类过氧化物酶催化)可导致氧化应激毒杀肿瘤;纳米材料催化H2O2歧化反应(类过氧化氢酶催化)可缓解乏氧抑制肿瘤增殖。因此,预测纳米材料对H2O2的催化活性可用于前瞻性地设计靶向H2O2的抗肿瘤纳米材料。高兴发课题组前期发展了预测纳米表面催化H2O2攫氢反应的理论模型(ACS Catal., 2020,10,12657),最近发展了预测纳米表面催化H2O2歧化反应的理论模型。这些模型基于催化机制,物理意义清晰,与密度泛函计算结合原则上可预测纳米材料的上述抗肿瘤活性。
为提高理论模型预测2D材料抗肿瘤活性的效率,进一步为模型中的描述符物理量训练了机器学习计算方法,建立了高效筛选抗肿瘤2D纳米材料的计算机方案。该方案兼具物理模型的可解释性与机器学习方法的高效性,用普通电脑与材料数据库连接即可实现虚拟筛选,为2D材料的抗肿瘤应用研究提供了高效理论工具(图1)。相关工作以Clear-box machine learning for virtual screening of 2D nanozymes to target tumor hydrogen peroxide为题,发表于Adv. Healthcare Mater. (论文链接:https://doi.org/10.1002/adhm.202202925),江西师范大学高雪皎副教授为第一作者,高兴发研究员为通讯作者。
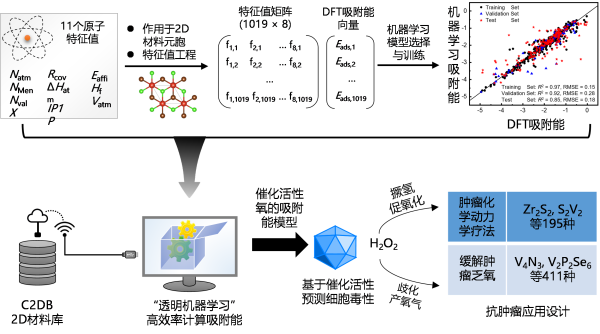
图1. 虚拟筛选抗肿瘤2D材料的“透明”机器学习方案。针对2D材料体系训练了计算H和OH吸附能的机器学习模型,将机器学习模型与预测催化活性的吸附能模型结合,获得了不依赖昂贵密度泛函计算,高效筛选潜在抗肿瘤材料的计算机方案。
2. 配体轨道能模型刻画MOF催化活性的远端取代基效应
纳米材料催化O2攫氢反应(类氧化酶催化)同样具有抗肿瘤应用前景,例如可利用该反应消耗肿瘤细胞的葡萄糖分子“饿死肿瘤”。前期实验发现MOF材料活化O2的活性与取代基的Hammett常数成正比,高兴发课题组通过密度泛函计算发现,MOF还原为该反应过程最可能的决速步,由于Hammett常数大的拉电子基团有利于MOF还原,可提高催化活性,解释了实验结果(Advanced Materials, 2021, 33, e2005024)。
然而,一些MOF在催化磷酸酯键水解时,推电子基团有利于提高催化活性。为深入研究MOF取代基调控催化活性的机制与规律,高兴发课题组以UiO-66为模型体系,研究了其催化两类反应的取代基效应,提出了“配体轨道能量模型”。该模型将催化反应的决速过渡态到前一个中间体的过程表示为决速过程,决速过程中MOF上的净电荷变化表示为ΔQMOF。模型指出,ΔQMOF的正负决定了取代基效应的方向性:当ΔQMOF > 0时,决速过程中电子从MOF向反应物转移,推电子基团有利于MOF给出电子,可提高催化活性;ΔQMOF < 0时,决速过程中电子从反应物向MOF转移,拉电子基团有利于MOF接受电子,可提高催化活性。由于取代基效应源自于连接配体π轨道与金属节点d轨道的远程π-d共轭,配体的LUMO(ELUMO)能级决定了取代基效应的作用强度。由此提出了图2所示的关系式定量描述取代基效应对MOF催化活性的调控规律。取代基效应一般用经典Hammett方程定量描述,而该方程使用经验参数,限制了其使用范围。配体轨道能量模型揭示了MOF催化取代基效应的底层机制,避免了经典Hammett方程使用不可移植经验参数的缺点,有望用于未来指导研制基于MOF孔道结构,具有反应底物选择性的纳米毒性调控技术。相关工作以Remote substituent effects on catalytic activity of metal-organic frameworks: a linker orbital energy model为题,发表于npj Comput. Mater. (论文链接:https://doi.org/10.1038/s41524-023-01008-5),国家纳米科学中心王真真副研究员为第一作者,高兴发研究员为通讯作者。
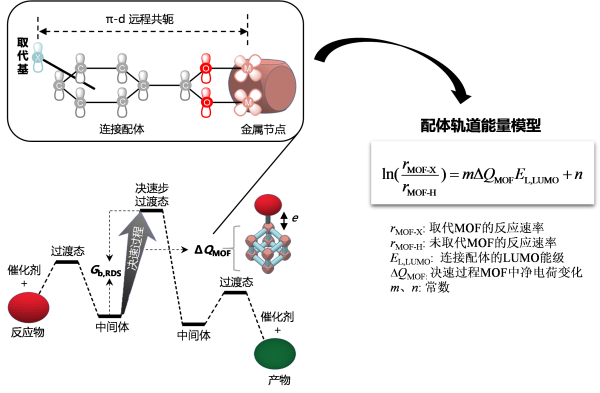
图2. 金属有机框架(MOF)催化活性取代基效应的机制与模型。根据该机制,反应决速过程中MOF上的静电荷变化ΔQMOF决定了取代基效应的方向性,连接配体LUMO能级ELUMO决定了取代基效应的强度。这揭示了MOF催化活性取代基效应的底层机制,避免了经典Hammett方程使用不可移植经验参数描述取代基效应的缺点。
3. 纳米酶催化机制与动力学的理论与计算研究
无机纳米材料催化H2O2攫氢、H2O2歧化、O2攫氢和O2??歧化的功能分别与氧化还原酶家族中过氧化物酶、过氧化氢酶、氧化酶和超氧化物歧化酶的催化功能相似,因此这些具有类酶催化活性的无机纳米材料也被称为纳米酶。纳米酶是材料、化学、生物和医学等学科交叉领域的热点研究方向之一。虽然纳米酶设计、合成、表征和应用研究已经取得了很大进展,但由于纳米酶工作环境复杂,其微观机制和动力学研究依然有挑战。密度泛函理论计算可在原子尺度刻画反应体系沿反应坐标方向的势能面,洞悉催化反应的微观机制和动力学,近年来在纳米酶催化机制研究中发挥了重要作用。这些计算结果或刻画纳米酶催化的原子历程,为实验研究提供支撑,或进一步发展理论模型描述催化活性,为计算设计提供理论工具。
高兴发研究员应董绍俊院士、阎锡蕴院士等人邀请为Advanced Materials期刊的《纳米酶专刊》撰写了题为Reaction mechanisms and kinetics of nanozymes: insights from theory and computation的综述,回顾了纳米材料模拟过氧化物酶、过氧化氢酶、氧化酶和超氧化物歧化酶等生物酶催化功能的分子机制和规律。重点评述了计算与实验如何交叉合作,共同揭示纳米酶催化机制,也展望了纳米酶计算和模拟研究未来面临的挑战性问题。相关工作发表于Adv. Mater. (论文链接:https://doi.org/10.1002/adma.202211151),江西师范大学沈小美博士为本文第一作者,高兴发研究员为通讯作者。
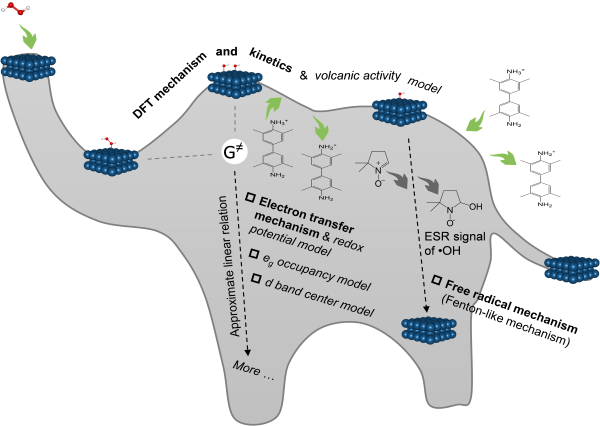
图3. 密度泛函理论(DFT)计算揭示纳米材料模拟过氧化物酶催化机制。该机制不涉及?OH,但可以解释实验中观察到“?OH信号”的原因,也可用于理解其他催化机制和预测模型。
4. “机制-理论-设计”指引发现其他靶向的纳米生物催化材料
纳米材料除与活性氧外发生催化作用外,与生物体内其他活性分子/基团的催化作用也是其毒性的来源。磷酸酯键是核酸、磷脂膜、蛋白质等生物结构中的重要官能团,为将化学模拟推广为一种研究纳米毒性上游化学机制的普适性策略,高兴发课题组研究了金属和金属氧化物表面水解磷酸酯键的微观机理和动力学,提出了“OH-H共吸附能模型”描述催化活性。与此前“路易斯酸模型”仅解释部分实验结果不同,新模型系统解释了前人实验,并预测了首例基于金属、稳定且生物相容度高的钌催化材料,用实验证实了预测结果(图4)。
该结果体现了“机制-理论-设计”这一化学模拟策略可作为一种研究纳米毒性上游化学理论与机制的普适性策略,研究复杂纳米生物体系中的化学问题。该研究成果以Mechanism and kinetics-guided discovery of nanometal scissors to cut phosphoester bonds为题,发表于ACS Catal. (论文链接:https://pubs.acs.org/doi/10.1021/acscatal.2c05094),国家纳米科学中心李巧枝博士和中科院生物物理所樊慧真博士为共同第一作者,高兴发研究员为通讯作者。
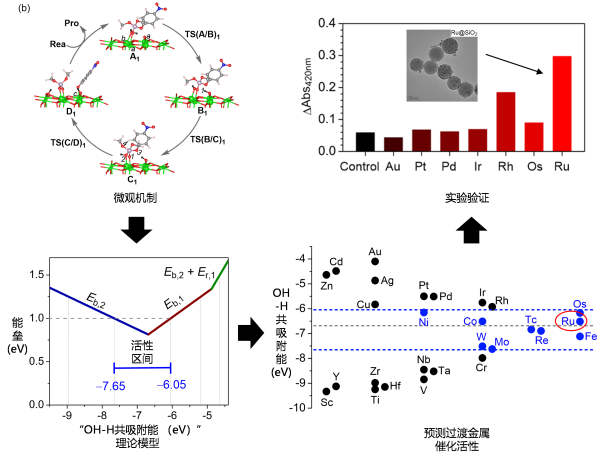
图4. 纳米材料催化水解磷酸酯键的机制、理论与设计。研究了金属和金属氧化物纳米表面催化磷酸酯键水解的微观机制,建立了理论模型,设计了金属钌催化材料,得到了实验验证。

