
近日,国家纳米科学中心任金东课题组在氮杂环亚胺分子与过渡金属表面吸附构型转变和界面电子转移研究方面取得新进展。相关成果以The electron-rich and nucleophilic N-heterocyclic imines on metal surfaces: binding modes and interfacial charge transfer为题,在线发表于Journal of the American Chemical Society(DOI: 10.1021/jacs.3c11738)。
功能分子材料是目前化学和材料学研究中的国际热点与前沿问题,也是当今纳米科技研究的前沿领域。以金属和有机功能分子材料为背景的研究及应用推动了材料科学、信息科学、能源及环境科学与技术等领域的进步。
任金东课题组长期从事功能分子表界面物性精准表征和操纵,通过精准调控分子中官能团空间构型,实现了卡宾表面分子构象的可逆翻转和可控自组装(Angew. Chem., Int. Ed. 61,e202115104(2022))。之后,在卡宾分子单体研究的基础上,基于扫描探针显微技术,探索并优化不同表面在位化学方法,克服位阻效应和表面分子热力学和动力学平衡等难题,首次在表面上实现了“Ballbot”型吸附的一维共价分子链的精准构筑和物性调控(Nat. Chem. 15, 1737–1744 (2023))。
氮杂环亚胺是一类基于Arduengo型氮杂环卡宾的亚胺配体,分子结构中包含的高度极化的环外碳氮双键,具有强供电子能力。由于其较卡宾增强的亲核性而被广泛用作分子表面锚定剂。此外,其强碱性和亲核性在催化有机合成反应中具有重要应用,其进一步表面功能化和催化应用亟需对其表面吸附构型和界面性质进行细致研究。近期,研究团队将STM、XPS和DFT等技术有机结合在一起,首次研究了不同过渡金属表面亲核氮杂化亚胺的表面结合和电学性质。咪唑环氮原子和亚胺氮原子的取代基通过改变分子表面构型影响其在表面直立或平躺的吸附模式,分子在不同金属基底的可公度性决定其表面结合模式及自组装分子层的致密程度。通过对覆盖度和退火参数的控制,实现了氮杂环亚胺分子从平躺到直立构型的转变。
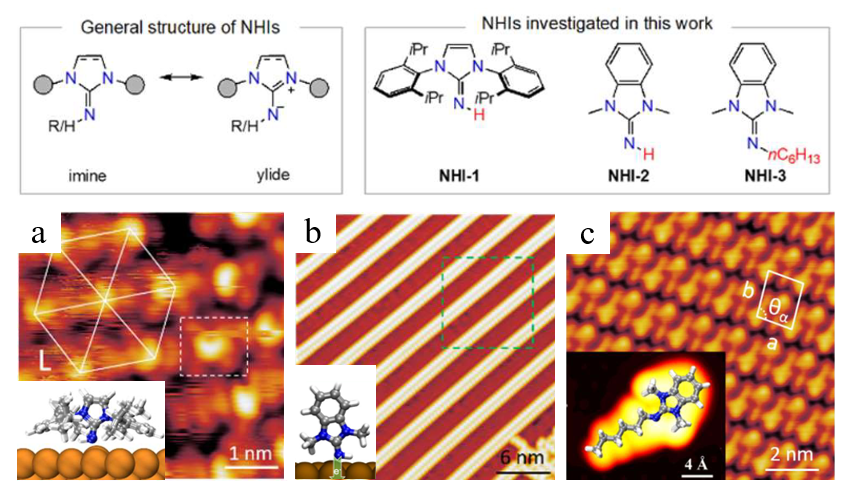
图1. Cu(111)表面的氮杂环亚胺结合模式与自组装。
进一步利用密度泛函理论,计算了亚胺单分子的表面电荷转移。分子表面吸附构型对其向金属表面的电荷注入效率具有显著影响,虽然平躺构型和具有较大氮取代基的亚胺分子单个分子的界面电子转移最高,具有较弱配体-金属相互作用的直立型分子体系,其直立分子链的堆积密度远远高于平躺型分子,因此是催化应用的最佳选择。
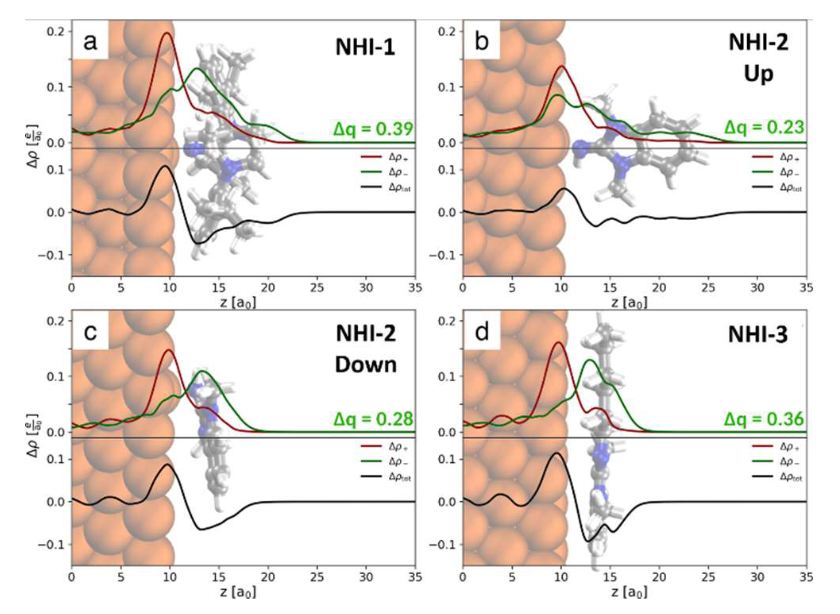
图2. Cu(111)表面的氮杂环亚胺差分电荷密度和转移电荷总量。
该工作为配位化学中非均相催化剂的设计优化提供了微观图像和分子层次的细致机理,在相关分子体系的催化和材料科学中的应用具有重要意义。
国家纳米科学中心为本工作第一单位,任金东研究员为本论文第一作者和通讯作者,德国明斯特大学博士研究生Mowpriya Das为共同第一作者,德国明斯特大学Saeed Amirjalayer教授、Harald Fuchs院士、Nikos L. Doltsinis教授、Frank Glorius院士为共同通讯作者。上述成果得到了国家重点研发计划、国家自然科学基金和中国科学院战略性先导科技专项等项目的资助。
论文链接:https://doi.org/10.1021/jacs.3c11738

